Supernova
Supernova is a software package for de novo assembly from Chromium Linked-Reads that are made from a single whole-genome library from an individual DNA source
Available Modules¶
module load Supernova/2.1.1Description¶
Supernova is a software package for de novo assembly from Chromium Linked-Reads that are made from a single whole-genome library from an individual DNA source. Supernova creates diploid assemblies, thus separately representing maternal and paternal chromosomes over long distances.
The Supernova software package includes two processing pipelines and one for post-processing:
supernova mkfastqwraps Illumina's bcl2fastq to correctly demultiplex Chromium-prepared sequencing samples and to convert barcode and read data to FASTQ files.supernova runtakes FASTQ files containing barcoded reads fromsupernova mkfastqand builds a graph-based assembly. The approach is to first build an assembly using read kmers (K = 48), then resolve this assembly using read pairs (to K = 200), then use barcodes to effectively resolve this assembly to K ≈ 100,000. The final step pulls apart homologous chromosomes into phase blocks, which are often several megabases in length.supernova mkoutputtakes Supernova's graph-based assemblies and produces several styles of FASTA suitable for downstream processing and analysis.
Download latest release from 10xGenomics.
https://support.10xgenomics.com/de-novo-assembly/software/downloads/latest
Availability¶
Supernova/2.1.1 is installed as a module and can be loaded via
module load Supernova
License¶
The developer grants a Limited License to all users. If you intend to use Supernova on NeSI operated infrastructure please read the developers own licensing agreement.
https://support.10xgenomics.com/de-novo-assembly/software/downloads/latest
Example script``¶
#SBATCH -J mySupernovajob
#SBATCH --partition=hugemem
#SBATCH --ntasks=1
#SBATCH --mem=460G
#SBATCH --cpus-per-task=16
#SBATCH --time=168:00:00
#SBATCH --hint=nomultithread
module load Supernova/2.1.1
supernova run --id=.....................
Getting Supernova to run successfully¶
We suggest users initially read the developers notes, at https://support.10xgenomics.com/de-novo-assembly/guidance/doc/achieving-success-with-de-novo-assembly
Further to that we also suggest,
- check --maxreads, to be passed to supernova, is correctly set. Recommended reading..https://bioinformatics.uconn.edu/genome-size-estimation-tutorial/http://qb.cshl.edu/genomescope/
- When passing
--localmemto supernova, ensure this number is less than the total memory passed to Slurm. - Pass
${SLURM_CPUS_PER_TASK}to supernova with the--localcoresargument.
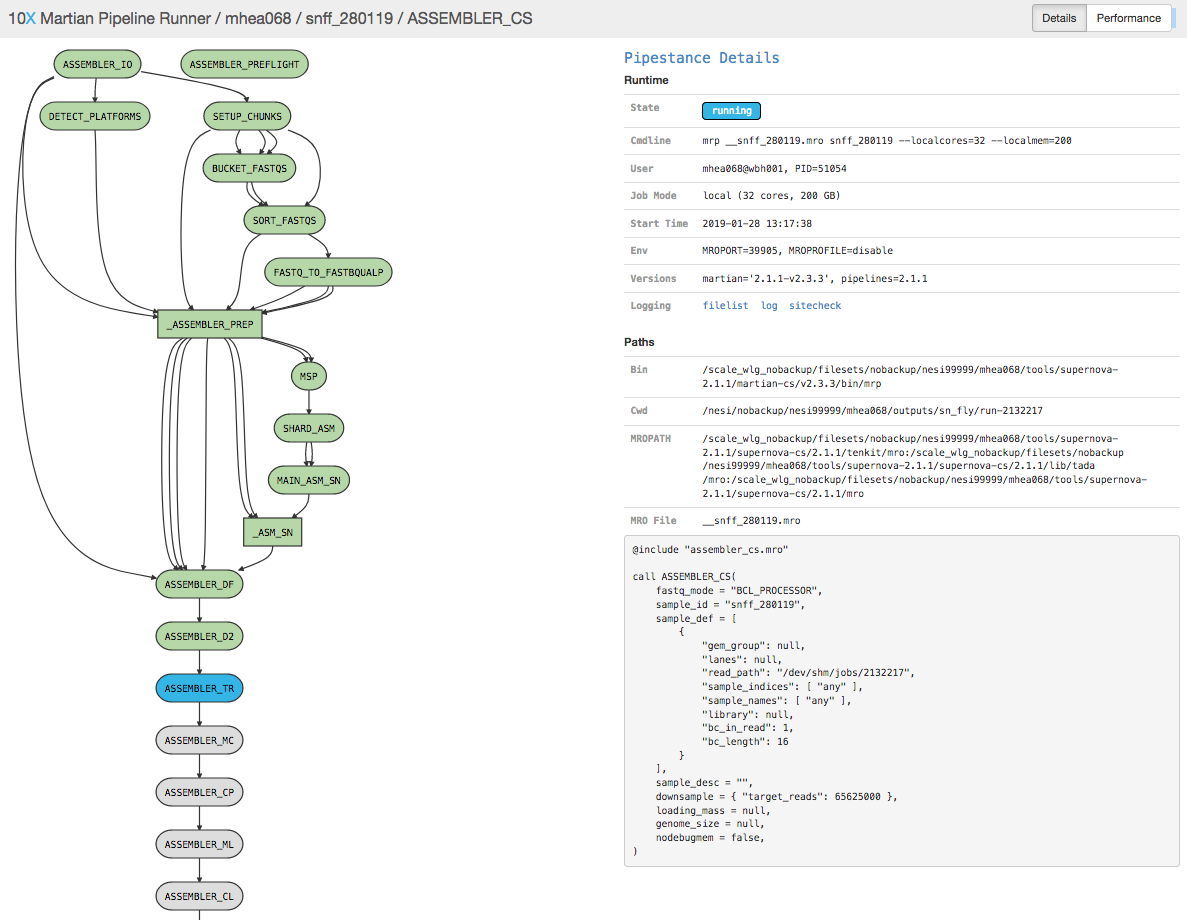
Tracking job progress via browser¶
Find the beginning of the _log file, located in the directory where
the call to supernova was run, or the path specified in the Slurm batch
file via --output.
head -n 30 <job_name>.out
supernova run (2.1.1)
Copyright (c) 2018 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Martian Runtime - '2.1.1-v2.3.3'
Serving UI at http://wbh001:37982?auth=Bx2ccMZmJxaIfRNBOZ_XO_mQd1njNGL3rZry_eNI1yU
Running preflight checks (please wait)...
Find the line..
Serving UI at http://wbh001:37982?auth=Bx2ccMZmJxaIfRNBOZ_XO_mQd1njNGL3rZry_eNI1yU
The link assumes the form..
http:// <node>: <port>?<auth>
- <node> Taken from above code snippet is wbh001
- <port> Taken from above code snippet is 37982
- <auth> Taken from above code snippet is Bx2ccMZmJxaIfRNBOZ_XO_mQd1njNGL3rZry_eNI1yU
In a new local terminal window open an ssh tunnel to the node. This takes the following general form
ssh -L <d>:<node>:<port> -N <server>
- <d> An integer
- <server> see: Standard Terminal Setup
When details are added to the general form from the specifics in the snippet above, the following could be run..
ssh -L 9999:wbh001:39782 -N mahuika
Next, open your preferred web browser and construct the following link..
http://localhost:<d>?<auth>
take <d> and <auth> from the code snippet above..
http://localhost:9999/?auth=Bx2ccMZmJxaIfRNBOZ_XO_mQd1njNGL3rZry_eNI1yU
Things to watch out for¶
- Supernova will create checkpoints after completing stages in the
pipeline. In order to run from a previously created checkpoint you
will first need to delete the _lock file located in the main output
directory (the directory named by
ID=${SLURM_JOB_NAME}where the_logfile is also located) and passed to supernova in the--id=${ID}argument in the sample Slurm script above. Avoid changing any other settings in both the call to Slurm and supernova.``````